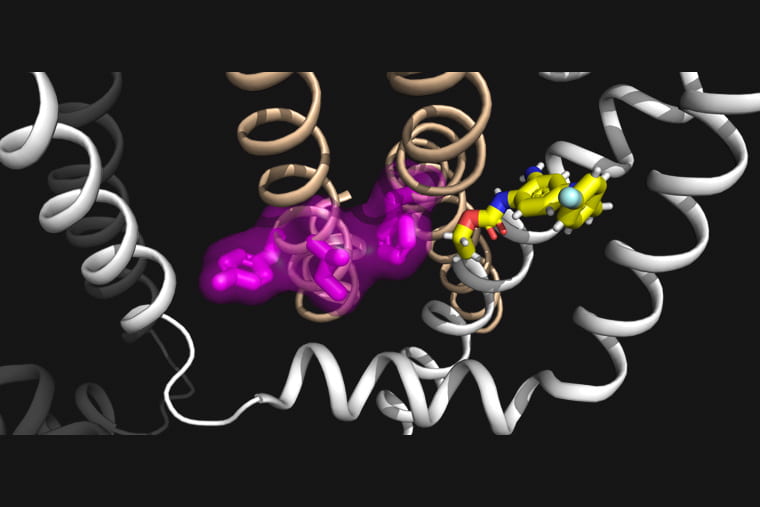
Epilepsy is a neurological disorder that arises from abnormal electrical activity in the brain leading to seizures. These seizure events can have a variety of causes, including genetic variants in a family of proteins that regulate potassium ions in the brain. Researchers at Washington University in St. Louis have led an international team to take a close look at the mechanisms behind the function and dysfunction of these proteins, as well as their interactions with an antiepileptic drug, to develop a potential new strategy to treat epilepsy.
Jianmin Cui, PhD, professor of biomedical engineering in the McKelvey School of Engineering, and Nien-Du Yang, a doctoral student in biomedical engineering who conducts research in Cui’s lab, teamed up with Harley Kurata, associate professor of pharmacology at the University of Alberta, and investigated the working mechanism of two potassium ion channels, KCNQ2 and KCNQ3. Their findings uncover a conserved mechanism for KCNQ channel activation that is a target of both epilepsy-linked mutations and a small molecule compound.
The work was published July 20 in Science Advances.
The KCNQ potassium channel family has multiple functions, from regulating heartbeat (by KCNQ1) to controlling excitability of neurons (by KCNQ2-5). These channels are voltage-activated so that they sense voltage changes across the cell membrane and open and close in response. The communication between voltage sensing and channel pore opening is known as electro-mechanical coupling, a process involving conformational changes of the protein during voltage-dependent activation.
Cui’s team has previously shown that KCNQ1, the cardiac KCNQ isoform, features a two-stage process in electro-mechanical coupling that leads to two distinct channel open states, the intermediate-open and activated-open. Regulation of the two open states underlies KCNQ1’s tissue-specific modulations, disease pathogenesis and pharmacology. KCNQ2 and KCNQ3 are highly expressed in the central nervous system and are the principal contributors to the M-current, a critical potassium current that modulates neuronal excitability. Therefore, impaired M-current function by congenital mutations in KCNQ2 and KCNQ3 is commonly associated with early-onset and pediatric epilepsy.